Sugar molecule helps stomach cells to differentiate between good and bad bugs
Max Planck scientists discover novel innate surveillance mechanism to fight off pathogenic Helicobacter strains
Half of all people are chronically infected with Helicobacter pylori, a Gram negative bacterium that plays a causative role in the development of gastric cancer. It comes in two types, one that is relatively harmless and another that increases the risk of cancer six-fold. The dangerous strain causes a particularly strong inflammation in the stomach mucosa by activating nuclear factor kappa B (NF-κB), the “master switch” of the tissue-based innate immune response, which rapidly triggers transcription of genes that initiate local inflammation. How exactly H. pylori triggers NF-κB has long been a mystery, however. Now a team at the Max Planck Institute for Infection Biology in Berlin has shown that the bacteria inject host cells with a sugar molecule, called HBP, found only in Gram negative bacteria, which activates NF-κB via a novel pathway. The researchers found that injection of HBP results in the rapid assembly of very large complexes, so-called TIFAsomes, which contain numerous proteins helping to orchestrate the anti-microbial inflammatory response in the infected stomach tissue.
 infected by Helicobacter pylori (green bacteria). TIFAsomes (red strings) formed upon injection of a small sugar molecule (HBP) by H. pylori into the host cells to cause translocation of the pro-inflammatory NF-kB transcription factor (diffused green) into the nuclei, as visualized in the right-side micrograph channel. Whitish/gray staining represents microtubule network of human cells (left panel).")
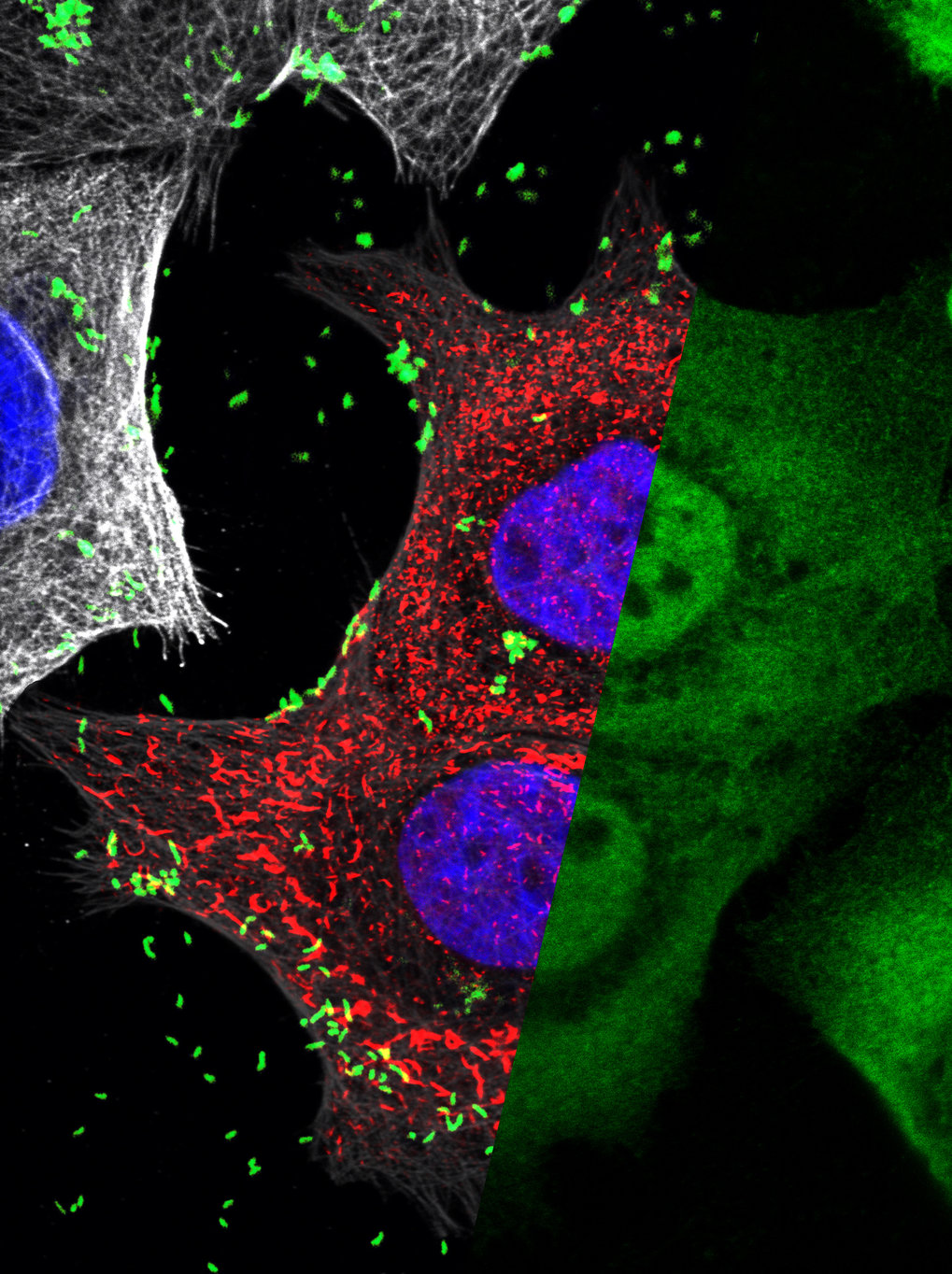
Chronic inflammation of the gastrointestinal tract is known to promote the development of cancer, thus understanding the mechanisms that initiate and control tissue-level inflammation is of great interest to human health. H. pylori, which causes life-long infections, sustains a constantly increased inflammatory response. This way, the pathogen can cause gastritis and stomach ulcers and was the first bacterium recognized to be carcinogenic. Interestingly, there are two major strains distinguished by the presence or absence of a type IV secretion system (T4SS) – a machinery used by the bacteria to inject molecules directly into the host cells, subverting their function. Strains which harbour the T4SS cause more severe inflammation and are more likely to cause ulcers and cancer – crucially, they are also the ones to strongly activate the master inflammatory regulator NF-κB.
How the cells of the stomach lining are able to distinguish these highly virulent from less virulent H. pylori has been the subject of intense research for decades. Until now, the injected virulence protein CagA, which triggers multiple deleterious changes in host cells, was the only molecule known to be translocated by H. pylori’s T4SS. Scientists naturally looked to this molecule to find the answer, but were unable to identify a clear mechanism that allowed the immune system to detect it.
A team led by Thomas F. Meyer at the Berlin-based Max Planck Institute for Infection Biology decided to take an unbiased genome-wide approach using RNAi interference technology and to examine which human genes were involved in activating NF-κB. “In particular, we were interested in genes that were activated after infection as compared to other inflammatory stimuli intrinsic to the stomach tissue. We suspected that the pathways involved were different.” In fact, the screen identified two unique molecules named ALPK1 and TIFA, which activated NF-κB only in response to H. pylori infection. Subsequent studies indicated that the ALPK1 kinase caused the phosphorylation of TIFA, which then triggered the rapid assembly of large protein complexes, called TIFAsomes. These complexes constituted the link to the classical NF-κB pathway. “When we used time-lapse microscopy of cells with fluorescently labelled TIFA, we were surprised at how rapidly these TIFA complexes were formed and how large they are,” says Prof Meyer. “We then realized just how many different proteins these TIFAsomes contain. Clearly they play a role in initiating other responses to infection, which we do not yet understand”.
Yet, the question remained how H. pylori sets in motion this chain of events. When another group found that HBP, a sugar molecule precursor of Gram-negative bacterial lipopolysaccharide synthesis, is capable of stimulating TIFAsome formation, the team tested if this sugar molecule could be the trigger in H. pylori NF-κB activation. Indeed, they observed that HBP from H. pylori could activate TIFA but for this H. pylori required a functioning T4SS. The realization that the delivery of HBP occurs via the virulence-associated T4SS came as a great surprise, as it was assumed to transfer only CagA. Since the T4SS has been recognized as the major determinant of H. pylori virulence, the fact that it specifically releases HBP explains how our body discriminates between highly pathogenic and less pathogenic strains. Only the highly pathogenic strains provoke a strong inflammation while our stomach mucosa remains quite tolerant towards the less pathogenic strains that do not have a T4SS.
RZ/HR